
팜이데일리 프리미엄 기사를 무단 전재·유포하는 행위는 불법이며 형사 처벌 대상입니다.
이에 대해 팜이데일리는 무관용 원칙을 적용해 강력히 대응합니다.
이에 대해 팜이데일리는 무관용 원칙을 적용해 강력히 대응합니다.
이 기사는 2025년4월30일 7시35분에 팜이데일리 프리미엄 콘텐츠로 선공개 되었습니다.
구독하기
[이데일리 송영두 기자] 제노스코 상장이 불발된 오스코텍이 신약개발 임상에서도 확실한 데이터를 내놓지 못하면서 후속 개발에 빨간불이 켜졌다. 진행성 고형암 치료제로 개발 중인 덴피본티닙 임상 1상 결과 안전성 우려가 제기됐기 때문이다. 회사 측은 안전성이 입증된 결과라고 주장하면서도 덴피본티닙 후속 개발 중단을 시사했다.
29일 이데일리 취재에 따르면 오스코텍(039200)은 진행성 고형암을 적응증으로 개발 중이던 AXL 저해제 덴피본티닙(SKI-G-801) 후속 개발을 중단할 것으로 확인됐다. 해당 후보물질은 최근 임상 1상 결과가 발표된 바 있다. 제노스코와 공동개발 중인 파이프라인으로, 기술이전 시 렉라자와 유사한 수익 모델이 될 수 있다는 점에서 주목을 받은 바 있다.
오스코텍 관계자는 “덴피본티닙 임상 1상 결과보고서(CSR)를 확인한 결과 안전성을 입증했다. 이번 임상 1상은 안전성과 내약성을 보는 시험으로 1a상을 진행했다. 안전성은 전혀 문제없는 수준으로 나왔다”면서도 “다음 단계 임상 개발은 하지 않는 쪽으로 무게를 두고 있다”고 말했다. 다만 CSR은 공개가 어렵다는 입장을 내놨다.
병용임상 필수인데...부작용 이슈↑, “후속 임상 신청시 허가 어려워”
지난 23일 오스코텍은 공시를 통해 AXL 저해제 덴피본티닙(SKI-G-801) 임상 1상 결과 보고서(CSR)를 수령했다고 밝혔다. 이번 임상 1상은 진행성 고형암 환자를 대상으로 덴피본티닙 단독요법의 안전성, 내약성 및 약동학적 특성을 평가하기 위한 공개, 다기관, 용량 증량 및 용량 결정으로 진행됐다. 목표 시험대상자 수는 36명이었지만, 21명만 등록됐다.
안전성 확인이 가장 중요했던 이번 임상 1상 결과 덴피본티닙을 한 번 이상 투여 받은 모든 시험대상자에서 총 105건의 이상사례가 발생했고, 이중 약물이상반응(ADR)은 61.90%였다. 21명중 13명에서 38건으로, 설사(diarrhea)와 메스꺼움(nausea)을 포함한 소화기계 이상사례가 빈번하게 발생했다.
특히 Grade 3 이상의 이상사례가 33.33%로 21명중 7명에서 14건이 발생했다. 중대한 이상사례도 14.29%로 21명 중 3명에서 4건, 중대한 약물이상반응은 9.52%로 21명 중 2명에서 3건이 발생했다. 중대한 약물 이상반응으로는 300㎎ 투약군에서 1건의 간도담계 이상반응으로 비혈액적 독성인 용량제한독성(DLT)에 해당했다. 해당 이상사례는 투약 영구 중단 후 폐암으로 진행해 시험대상자가 사망했다.
300㎎ 투약군에서는 심전도 이상 사례 1건이 발생해 투약이 중단됐고, 용량을 늘린 400㎎ 투약군에서도 Grade 3가 1건 발생했는데 DLT로 확인됐다. 후속 임상 용량은 400㎎으로 설정했다. 리가켐바이오(141080)가 중국 시스톤사에 기술이전해 고형암 치료제로 개발되고 있는 LCB71의 경우 표적 타깃은 다르지만 지난해 8월 발표된 임상 1a상 결과 용량제한독성(DLT)은 관찰되지 않았다. 관찰된 치료 관련 부작용도 Grade 1 또는 2가 대부분으로 Grade 3 이상의 중증 부작용은 14.3%에 그쳐 충분한 내약성이 확인됐다는 평가를 받은 바 있다.
반면 오스코텍의 이번 임상 1상 결과를 확인한 업계와 전문가들은 안전성에 우려를 표했다. 항암제 개발사 임상 연구팀 관계자는 “저분자의약품(스몰몰레큘)으로 개발시 간독성과 심장독성에 굉장히 예민한데, 심장 독성이 발생했다는 것은 간과할 수 없는 부분”이라면서 “300㎎에서 심장 독성이 나왔는데, 후속 임상을 400㎎으로 올려 진행하겠다는 것은 무리수라고 판단된다”고 말했다.
글로벌 CRO(임상시험수탁기관) 출신 전문가는 “용량제한독성이 1건씩 나와서 안전성 범주에 해당한다고 표현한 것 같은데, Grade 3 부작용 비율도 높다. 중증 이상반응 비율도 높아서 상당히 부작용이 높은 편에 속한다”면서 “안전성은 기본 중의 기본인데, 이대로 후속 임상 개발은 어려울 가능성이 높다. 규제기관에서 추가 실험 등을 통해 안전성 입증을 요구할 것이다. 후속 임상시험계획 신청(IND)을 안 받아줄 가능성이 농후하다”고 강조했다.
특히 항암제 임상 특성상 병용임상이 대부분인 만큼 병용임상시 부작용 이슈는 더욱 커질 수 있을 것이란 분석도 나온다. 실제로 오스코텍 측이 덴피본티닙 후속 임상인 1b/2a상을 병용임상으로 진행하려 했었던 것으로 확인됐다.
임상 연구 전문가는 “항암제는 단독요법보다 병용요법 데이터가 훨씬 잘 나오는 만큼 최근 트렌드는 병용요법이다. 하지만 단독요법에서 부작용 이슈가 있는데, 병용임상시에는 부작용 이슈가 더욱 커질 수밖에 없다”면서 “회사가 직접 약물을 사서 병용임상을 진행하는 것이라면 모를까 다른 기업에 병용 임상을 제안하기 어려울 것이다. 제안하더라도 같이 공동 개발하려고 나서는 기업은 찾기 쉽지 않을 수 있다”고 설명했다.
임상 성공했다면서, 개발 중단...왜?
오스코텍 측은 안전성 우려가 제기되는 것과 관련해서 반복해서 안전성이 입증된 것이라고 강조했다. 공시를 통해서도 “Grade 3 이상의 비혈액학적 독성 및 혈액학적 독성이 일부 보고됐으나, 위장관계 독성 및 혈액학적 독성을 포함한 이상사례 전반적으로 기존에 보고된 AXL 억제제 계열 약물의 안전성 데이터와 유사한 양상을 보였고, 임상적으로 수용 가능한 수준으로 판단된다”고 평가했다.
즉 임상 1상에서 안전성을 입증했다는 설명인데, 임상 성공에도 불구하고 회사 측은 덴피본티닙 후속 개발을 하지 않기로 한 이유에 대해서는 전략적인 결정이라고 강조했다.
오스코텍 관계자는 “덴피본티닙은 후속 임상인 1b/2상은 전략적으로 하지 않을 것이다. 또 다른 파이프라인 OCT-598도 고형암으로 개발하고 있고, 항 내성제이지만 겹치는 부분이 있다”며 “덴피본티닙이 속한 고형암 치료제 시장은 경쟁자가 많아 확실한 경쟁력을 갖기 어렵겠다는 부분을 내부적으로 인지하고 있다”고 설명했다.
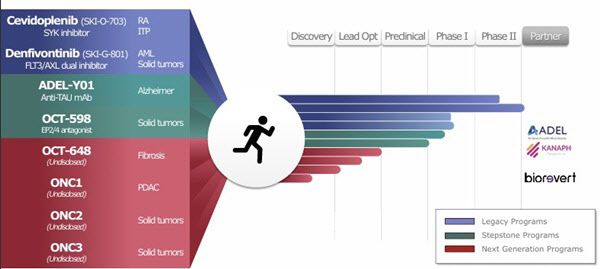
오스코텍의 이번 덴피본티닙 임상 1상 결과는 아쉬울 수밖에 없다는 게 업계 평가다. 렉라자 이후 제2 렉라자 출현이 절실한 시점에서 가장 개발 속도가 빠르고, 큰 잠재력을 갖췄다고 평가받았던 세비도플레닙은 △류머티즘관절염 미국 임상 2상 실패 △면역혈소판감소증 미국 임상 2상 실패로 사실상 동력을 잃었다. 여기에 두 번째로 빠른 개발이 이뤄졌던 덴피본티닙의 1상 결과와 후속 개발 자진 중단은 오스코텍과 제노스코의 신약개발 신뢰도와 능력에 타격을 줄 수 있다는 분석이다.
세비도플레닙과 데피본티닙 모두 오스코텍과 제노스코가 공동 개발하던 파이프라인이라는 점에서 오스코텍은 물론 기술성 평가에서 업계 최초로 AA, AA를 받았던 제노스코에 대한 재평가도 이뤄져야 한다는 지적도 나온다. 현재 오스코텍 파이프라인 중 아델사와 공동개발 중인 알츠하이머 치료제(미국 임상 1상)를 제외하면 5개 파이프라인 모두 전임상 단계다.
29일 이데일리 취재에 따르면 오스코텍(039200)은 진행성 고형암을 적응증으로 개발 중이던 AXL 저해제 덴피본티닙(SKI-G-801) 후속 개발을 중단할 것으로 확인됐다. 해당 후보물질은 최근 임상 1상 결과가 발표된 바 있다. 제노스코와 공동개발 중인 파이프라인으로, 기술이전 시 렉라자와 유사한 수익 모델이 될 수 있다는 점에서 주목을 받은 바 있다.
오스코텍 관계자는 “덴피본티닙 임상 1상 결과보고서(CSR)를 확인한 결과 안전성을 입증했다. 이번 임상 1상은 안전성과 내약성을 보는 시험으로 1a상을 진행했다. 안전성은 전혀 문제없는 수준으로 나왔다”면서도 “다음 단계 임상 개발은 하지 않는 쪽으로 무게를 두고 있다”고 말했다. 다만 CSR은 공개가 어렵다는 입장을 내놨다.
|
병용임상 필수인데...부작용 이슈↑, “후속 임상 신청시 허가 어려워”
지난 23일 오스코텍은 공시를 통해 AXL 저해제 덴피본티닙(SKI-G-801) 임상 1상 결과 보고서(CSR)를 수령했다고 밝혔다. 이번 임상 1상은 진행성 고형암 환자를 대상으로 덴피본티닙 단독요법의 안전성, 내약성 및 약동학적 특성을 평가하기 위한 공개, 다기관, 용량 증량 및 용량 결정으로 진행됐다. 목표 시험대상자 수는 36명이었지만, 21명만 등록됐다.
안전성 확인이 가장 중요했던 이번 임상 1상 결과 덴피본티닙을 한 번 이상 투여 받은 모든 시험대상자에서 총 105건의 이상사례가 발생했고, 이중 약물이상반응(ADR)은 61.90%였다. 21명중 13명에서 38건으로, 설사(diarrhea)와 메스꺼움(nausea)을 포함한 소화기계 이상사례가 빈번하게 발생했다.
특히 Grade 3 이상의 이상사례가 33.33%로 21명중 7명에서 14건이 발생했다. 중대한 이상사례도 14.29%로 21명 중 3명에서 4건, 중대한 약물이상반응은 9.52%로 21명 중 2명에서 3건이 발생했다. 중대한 약물 이상반응으로는 300㎎ 투약군에서 1건의 간도담계 이상반응으로 비혈액적 독성인 용량제한독성(DLT)에 해당했다. 해당 이상사례는 투약 영구 중단 후 폐암으로 진행해 시험대상자가 사망했다.
300㎎ 투약군에서는 심전도 이상 사례 1건이 발생해 투약이 중단됐고, 용량을 늘린 400㎎ 투약군에서도 Grade 3가 1건 발생했는데 DLT로 확인됐다. 후속 임상 용량은 400㎎으로 설정했다. 리가켐바이오(141080)가 중국 시스톤사에 기술이전해 고형암 치료제로 개발되고 있는 LCB71의 경우 표적 타깃은 다르지만 지난해 8월 발표된 임상 1a상 결과 용량제한독성(DLT)은 관찰되지 않았다. 관찰된 치료 관련 부작용도 Grade 1 또는 2가 대부분으로 Grade 3 이상의 중증 부작용은 14.3%에 그쳐 충분한 내약성이 확인됐다는 평가를 받은 바 있다.
반면 오스코텍의 이번 임상 1상 결과를 확인한 업계와 전문가들은 안전성에 우려를 표했다. 항암제 개발사 임상 연구팀 관계자는 “저분자의약품(스몰몰레큘)으로 개발시 간독성과 심장독성에 굉장히 예민한데, 심장 독성이 발생했다는 것은 간과할 수 없는 부분”이라면서 “300㎎에서 심장 독성이 나왔는데, 후속 임상을 400㎎으로 올려 진행하겠다는 것은 무리수라고 판단된다”고 말했다.
글로벌 CRO(임상시험수탁기관) 출신 전문가는 “용량제한독성이 1건씩 나와서 안전성 범주에 해당한다고 표현한 것 같은데, Grade 3 부작용 비율도 높다. 중증 이상반응 비율도 높아서 상당히 부작용이 높은 편에 속한다”면서 “안전성은 기본 중의 기본인데, 이대로 후속 임상 개발은 어려울 가능성이 높다. 규제기관에서 추가 실험 등을 통해 안전성 입증을 요구할 것이다. 후속 임상시험계획 신청(IND)을 안 받아줄 가능성이 농후하다”고 강조했다.
특히 항암제 임상 특성상 병용임상이 대부분인 만큼 병용임상시 부작용 이슈는 더욱 커질 수 있을 것이란 분석도 나온다. 실제로 오스코텍 측이 덴피본티닙 후속 임상인 1b/2a상을 병용임상으로 진행하려 했었던 것으로 확인됐다.
임상 연구 전문가는 “항암제는 단독요법보다 병용요법 데이터가 훨씬 잘 나오는 만큼 최근 트렌드는 병용요법이다. 하지만 단독요법에서 부작용 이슈가 있는데, 병용임상시에는 부작용 이슈가 더욱 커질 수밖에 없다”면서 “회사가 직접 약물을 사서 병용임상을 진행하는 것이라면 모를까 다른 기업에 병용 임상을 제안하기 어려울 것이다. 제안하더라도 같이 공동 개발하려고 나서는 기업은 찾기 쉽지 않을 수 있다”고 설명했다.
임상 성공했다면서, 개발 중단...왜?
오스코텍 측은 안전성 우려가 제기되는 것과 관련해서 반복해서 안전성이 입증된 것이라고 강조했다. 공시를 통해서도 “Grade 3 이상의 비혈액학적 독성 및 혈액학적 독성이 일부 보고됐으나, 위장관계 독성 및 혈액학적 독성을 포함한 이상사례 전반적으로 기존에 보고된 AXL 억제제 계열 약물의 안전성 데이터와 유사한 양상을 보였고, 임상적으로 수용 가능한 수준으로 판단된다”고 평가했다.
즉 임상 1상에서 안전성을 입증했다는 설명인데, 임상 성공에도 불구하고 회사 측은 덴피본티닙 후속 개발을 하지 않기로 한 이유에 대해서는 전략적인 결정이라고 강조했다.
오스코텍 관계자는 “덴피본티닙은 후속 임상인 1b/2상은 전략적으로 하지 않을 것이다. 또 다른 파이프라인 OCT-598도 고형암으로 개발하고 있고, 항 내성제이지만 겹치는 부분이 있다”며 “덴피본티닙이 속한 고형암 치료제 시장은 경쟁자가 많아 확실한 경쟁력을 갖기 어렵겠다는 부분을 내부적으로 인지하고 있다”고 설명했다.
오스코텍의 이번 덴피본티닙 임상 1상 결과는 아쉬울 수밖에 없다는 게 업계 평가다. 렉라자 이후 제2 렉라자 출현이 절실한 시점에서 가장 개발 속도가 빠르고, 큰 잠재력을 갖췄다고 평가받았던 세비도플레닙은 △류머티즘관절염 미국 임상 2상 실패 △면역혈소판감소증 미국 임상 2상 실패로 사실상 동력을 잃었다. 여기에 두 번째로 빠른 개발이 이뤄졌던 덴피본티닙의 1상 결과와 후속 개발 자진 중단은 오스코텍과 제노스코의 신약개발 신뢰도와 능력에 타격을 줄 수 있다는 분석이다.
세비도플레닙과 데피본티닙 모두 오스코텍과 제노스코가 공동 개발하던 파이프라인이라는 점에서 오스코텍은 물론 기술성 평가에서 업계 최초로 AA, AA를 받았던 제노스코에 대한 재평가도 이뤄져야 한다는 지적도 나온다. 현재 오스코텍 파이프라인 중 아델사와 공동개발 중인 알츠하이머 치료제(미국 임상 1상)를 제외하면 5개 파이프라인 모두 전임상 단계다.
송영두 songzio@


함께보면 좋은 뉴스



![지놈앤컴퍼니, 임상 본격화 기대감에 급등…숨고르기 들어간 바이젠셀[바이오 맥짚기]](https://image.edaily.co.kr/images/vision/files/NP/S/2026/04/PS26043000447b.jpg)
